CANCER IMMUNITY
A Critical Concept to Understanding Carcinogenesis

Abnormalities of the immune system play a critical role in the development of cancers with a breakdown in immune surveillance, abnormalities of cell mediated immunity, angiogenesis together with excessive inflammation. Restoring the immune system is an essential component of the Metabolic Approach to treating cancer. … its part of the HEALING process. The use of conventional chemotherapy would appear to be counter-intuitive at it destroys the immune system. - Paul.

The Tumor Microenvironment
The tumor microenvironment is the complex ecosystem of cells, molecules, and structures that surround and interact with tumor cells, and it plays an active role in how cancers grow, spread, and respond to treatment.(1-5) It is not just a passive background but a dynamic partner in cancer progression that tumors actively reshape to support their own survival.
Main components
The tumor microenvironment contains many non-cancerous cell types that communicate continually with tumor cells. Key components include (see figure 1).
Immune cells such as T cells, B cells, natural killer cells, macrophages, and others that can either attack or support the tumor.
Stromal cells, especially cancer‑associated fibroblasts (CAFs), which produce structural proteins and growth factors that help remodel tissue around the tumor.
Blood vessels and endothelial cells that supply oxygen and nutrients, along with pericytes that stabilize these vessels.
Extracellular matrix (ECM), the structural “scaffolding” of proteins and fibers that gives tissues shape and influences how cells move and behave.
Soluble signaling molecules, including cytokines, chemokines, and growth factors that coordinate communication among all these cells.
How tumors reshape the environment
As tumors grow, they actively reprogram their surroundings to favor cancer cell survival and immune evasion. They do this by:
Releasing signals that convert normal fibroblasts into cancer‑associated fibroblasts, which then promote proliferation, invasion, and new blood vessel growth.
Attracting and polarizing immune cells, such as macrophages, toward tumor‑supporting states that suppress effective anti‑tumor immunity.
Inducing abnormal, leaky blood vessels that can both feed the tumor and hinder efficient delivery of drugs.
Building physical and biochemical barriers in the ECM that make it harder for immune cells and therapies to penetrate the tumor mass.
Metabolic and physical conditions
The tumor microenvironment often has distinct metabolic and physical features that further influence cancer behavior.
Hypoxia (low oxygen) develops because rapidly growing tumors outstrip their blood supply, which in turn drives new vessel formation and can make cancer cells more aggressive and treatment‑resistant.
Acidity increases as cancer cells rely heavily on glycolysis and produce lactic acid, creating an environment that impairs anti‑tumor immune cells but favors certain tumor‑promoting immune populations.
Increased matrix stiffness and abnormal tissue architecture can enhance cancer cell migration and invasion into surrounding tissues.
Clinical significance
Because the tumor microenvironment shapes how cancers grow and respond to therapy, it has become a major focus of modern cancer treatment strategies.
Immunotherapies aim to “re-educate” immune cells in the microenvironment or block suppressive signals so that immune responses against the tumor are restored.
Anti‑angiogenic drugs target abnormal tumor blood vessels to limit nutrient supply and normalize vessel structure.
Emerging therapies specifically target cancer‑associated fibroblasts, ECM components, or key signaling molecules in the microenvironment to disrupt the support system that tumors depend on.
Inflammation is an essential pillar of immune defense. However, chronic inflammation is considered a hallmark of cancer initiation and progression. Chronic inflammation demonstrates a potential to induce complex changes at molecular, cellular, and organ level and thereby alter the tumor microenvironment (TME). Cancer cells frequently secrete several growth factors that stimulate myelopoiesis and recruit myeloid cells to TME (see Figure 1). (4, 6) Therefore, the TMEs of various cancers are characterized by the high infiltration of monocytes, macrophages, granulocytes, and dendritic cells. Most myeloid cells within TMEs are present in an immature form; however, cancer-derived growth factors modify these myeloid cells into cells that support carcinogenesis by enhancing proliferation, migration, and metastasis and enabling cancer cell survival and immune evasion. Therefore, in addition to abnormalities in apoptosis, patients with cancer have derangements in immunity with the immune system failing to recognize the cancer cell as foreign. The following cells play a major role in altering the TME and promoting carcinogenesis.
Figure 1. Cancer microenvironment
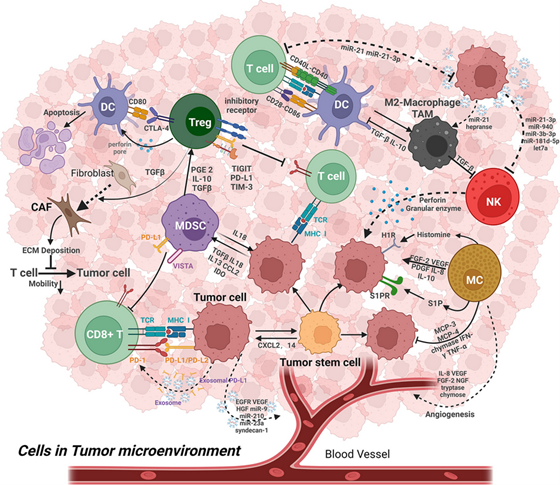
Myeloid-derived suppressor cells (MDSC). The establishment of primary tumor cells in distant organs, termed metastasis, is the principal cause of cancer mortality. Despite “curative” resection of the primary tumor, many patients have disseminated tumor cells at the time of diagnosis. Tumor cells can be found in the bone marrow of cancer patients at the time of their primary tumor resection. (7) These patients may then develop overt metastases months, years, or even decades later. This latency period, during which cancer cells do not grow and remain in a quiescent or equilibrium state, is known as “cancer dormancy.” The timeline of metastatic dormancy is regulated by interactions between the tumor, its microenvironment, angiogenesis, and tumor antigen-specific T-cell responses. One such mediator of dormancy is myeloid-derived suppressor cells (MDSCs), whose number in infiltrating tumors has been associated with cancer stage, grade, patient survival, and metastasis in a broad range of tumor pathologies (see Figure 4). (8, 9)
Extensive studies have revealed a role for MDSCs in tumor escape from adaptive and innate immune responses, facilitating tumor progression and metastasis. (8, 10-14) Host immunity via tumor-specific cytotoxic T-lymphocytes can control disseminated tumor cell growth,
resulting in a dormant lesion that can be held in stasis for years or decades until released from dormancy in association with an increase in MDSCs reversing host T-cell responses. MDSCs contribute to immune evasion by inducing T-cell dysfunction through the production of reactive oxygen species, arginase-1 (ARG1), and nitric oxide synthase (NOS). ARG1 hydrolyzes extracellular L-arginine into urea and ornithine. L-arginine is required for T-cell proliferation, cytokine production, and expression of the T-cell receptor. (15)
Figure 2. Crosstalk between MDSCs and other immune cells. (9)
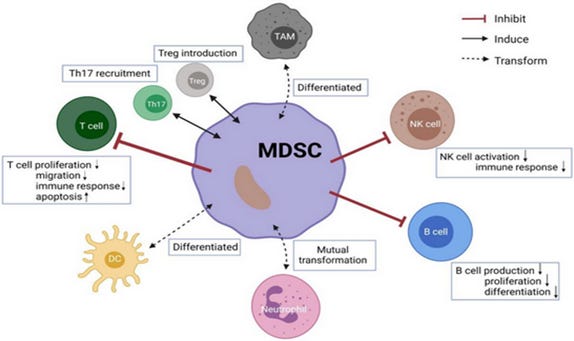
MDSCs can not only inhibit clonal expansion of activated effector T cells, but also induce tumor-specific Treg lymphocytes to further establish and maintain T-cell tolerance in the tumor-bearing host. (13, 16, 17) In addition, by downregulating interferon, overexpressing inflammatory cytokines, and creating leaky vasculature by overexpressing matrix metalloproteinase 9 and other remodeling factors which compromise the integrity of the extracellular matrix and the basal membrane, MDSCs promote cancer cell invasion. (12)
T-regulatory cells (Tregs). Tregs universally labeled by CD4+CD25+Foxp3+CD127low/− are differentiated from traditional T lymphocytes. (18-21) To maintain immune homeostasis, Treg cells inhibit abnormal or excessive immune reactions to self- and non-self-antigens. By stifling the anti-tumor immune response of effector T cells, NK cells, and dendritic cells, Treg cells contribute to the growth and spread of tumors in the TME. (19, 21, 22) An unfavorable prognosis is associated with high Treg cell infiltration in the TME in patients with diverse cancer types. (22-28) Treg cells cause immune suppression by the production of immunosuppressive cytokines, the consumption of interleukin-2 and IL 2 receptors, modulation of CD80 and CD86 expression by dendritic cells, and direct killing of effector T cells. (22) Tregs also contribute significantly to angiogenesis via the VEGF/VEGFR pathway.
Natural Killer Cells (NK cells). Natural killer (NK) cells are the most relevant cancer-fighting cells of the innate immune system. NK cells play a vital role in recognizing and responding to abnormal cells, including cancerous and infected cells, in the immune system. T-cells possess T-cell receptors (TCRs) that allow them to bind MHC-I-peptide complexes on the cell surface, which determines whether an immune response will be initiated. Failures occur in the expression of the transporter associated with antigen processing (TAP) complex, and β2-microglobulin; these cause a loss of MHC-I self-antigen transport and surface presentation capacity, which causes the failure of the NK cell to destroy the cancerous cell.
Tumor-associated macrophages. Macrophages recruited from circulating monocytes to tumors and influenced by the presence of cancer to promote tumor malignancy and progression are often referred to as tumor-associated macrophages (TAMs) (see Figure 5). (29-31) Macrophages are divided into the M1 and M2 subgroups based on morphological, phenotypic, and functional variability. M2 macrophages have been shown to have protumor characteristics and to promote tumor development and metastasis, whereas M1 macrophages play a critical role in antitumor immunity and largely mediate proinflammatory activities in the tumor microenvironment (TME). (31-33) In metastatic tumors, macrophages have different phenotypes and functions from primary tumors and are often called metastasis-associated macrophages (MAMs).
TAMs mostly arise from bone-marrow-derived monocytes with the chemokine CCL2 produced by tumor cells being the major recruitment factor. Bone-marrow-derived monocytes include both classical monocytes and monocytic MDSCs (M-MDSCs), (34) and are crucial for the negative regulation of immune responses. (5, 35)
The immune system is skewed toward a tumor-promoting response because of the release of IL-10 by MDSCs, which inhibits the secretion of IL-12 by macrophages. Macrophages also cause MDSCs to produce more IL-10, which raises levels of IL-6 and TNF- in macrophages. (5) MDSC IL-6 was reported to be elevated by tumor cells, and vice versa. (5) The ratio of tumor cells to MDSC and macrophages controls inflammation within solid tumors, and interactions between these cells have the potential to drastically change the inflammatory environment within the tumor microenvironment. (5, 32) A high infiltration of macrophages in human solid tumors is associated with poor clinical outcomes. (5, 31-33, 35-43) Similarly, the expression of macrophage growth factors or their chemoattractants, such as CSF1 and CCL2, in tumors or in the circulation is often associated with poor prognosis. (29)
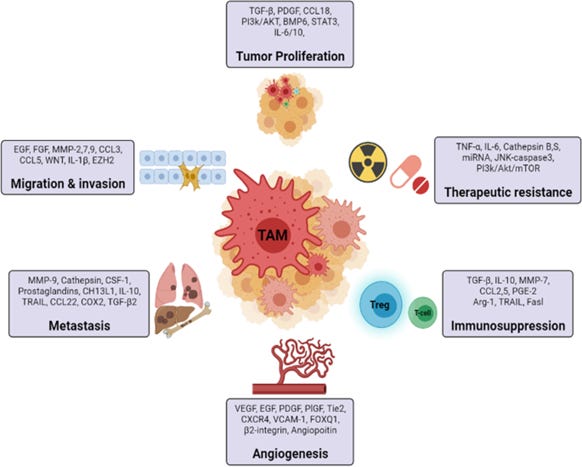
TAMs are the crucial and dominant immune cells in the TME and significantly contribute to tumor progression by promoting angiogenesis, mediating tumor immunosuppression by inhibiting T cell function, they secrete chemokines which contribute to the recruitment of T regulatory cells in the tumor microenvironment and promote tumor cell intravasation via VEGF expression (see Figure 5). TAMs are activated by mediators secreted from tumor-infiltrating lymphocytes such as Th2, Treg cells, IL-10, TGF-β. (44) By reducing antitumor immunity, Foxp3+ regulatory T (Treg) cells and tumor-associated macrophages (TAMs) both aid in the growth of tumors. Researchers identified TAMs and Tregs as responsible for direct tumor immune evasion. (45) TAMs and Tregs combine to form a cellular network that is partially redundant and contributes to the robustness of tumor immunosuppression as well as resistance to immunotherapy. (32, 45)
TAMs play a major role in tumor metastases. (46) Cancer-associated fibroblast are produced because of the mesenchymal transition of endothelial cells during the growth of tumors, and they secrete Heat shock protein-90 alpha (Hsp90α), which promotes M2 polarization and maintains an immunosuppressive milieu. (47) By secreting different mediators that alter the tumor promoting TME, TAMs can accelerate the growth of tumors. Proangiogenic growth factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor
(PDGF), fibroblast growth factor (FGF), and transforming growth factor- (TGF-), NF-kB-mediated factors that prevent apoptosis, and proangiogenic growth factors (38) that promote cancer cell migration and metastasis. (32) TAMs can also increase the tumor stemness, which upregulates the release of immunosuppressive cytokines such as IL-1ra. (32, 48) By releasing growth factors like the epidermal growth factor receptor (EGFR), which encourages the proliferation of cancer cells, TAMs may directly drive the proliferation of cancer cells. (49) In hepatocellular carcinoma, active Wnt/-catenin signaling induced by a greater number of invading macrophages can promote the proliferation of tumor progenitor cells, and targeted macrophage reduction can diminish Wnt and slow tumor growth. (50)
By controlling the PI3k/Akt pathways in cancer cells, TAMs may block proapoptotic cytokines such tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). (51) By introducing miRNAs into cancer cells, such as colorectal cancer and pancreatic ductal adenocarcinoma cells, exosomes produced by M2 macrophages spread malignancy. (52) Metastatic cells use the Cysteine-cysteine motif chemokine ligand 20 (CCL20), also known as macrophage inflammatory protein-3α, MIP3α) - Chemokine receptor 6 (CCR6) axis/pathway to attract monocytes and differentiate them into metastasis-associated macrophages (MAMs) that support tumor cell survival and metastasis by suppressing T cells. (32, 37) Additionally, TAMs release several enzymes, such as matrix metalloproteinases (MMPs) and cyclooxygenase type-2 (COX-2), which all work to promote angiogenesis by destroying the matrix and enabling endothelial cells to invade. (53) Despite TAMs having pro-tumorigenic characteristics, they can ingest tumor cells, and cause tumor apoptosis by releasing NO, ROS, and IL-12, which encourage anti-tumor responses and limit tumor growth in specific situations. (54) This suggests that immunosuppressive and immunostimulatory TAM can coexist in the same tumor. (5, 32, 55)
Figure 3. The role of tumor associated macrophages in cancer. (31)
PLATELETS AND CANCER
Platelets have been implicated in enabling successful metastasis and worsening the prognosis of patients with cancer by guarding tumor cells from immune elimination and promoting arrest and extravasation of tumor cells. (56-58) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Platelet-derived TGFβ and direct platelet-tumor cell contacts synergistically activate the TGFβ/Smad and NF-κB pathways in cancer cells, resulting in their transition to an invasive mesenchymal-like phenotype and enhanced metastasis in vivo. (56) In a symbiotic manner, tumor-derived bioactive molecules have been shown to prompt an increase in platelet activation and production. (59, 60)
ANGIOGENESIS AND METASTASIS
Angiogenesis involves neovascularization or the formation of new capillaries from existing blood vessels and is associated with the processes of tissue inflammation, wound healing, and tumorigenesis. Angiogenesis is required for most tumors to grow beyond an approximate size of 0.2-2.0 mm. In addition to its role in up-regulating glycolysis in response to hypoxia, HIF-1α is the main transcription factor for vascular endothelial growth factor (VEGF), which stimulates angiogenesis.
Metastasis is the general term used to describe the spread of cancer cells from the primary tumor to surrounding tissues and to distant organs and is a primary cause of cancer morbidity and mortality. To complete the metastatic cascade, cancer cells must detach from the primary tumor, intravasate into the circulatory and lymphatic systems, evade immune attack, extravasate at distant capillary beds, and invade and proliferate in distant organs. The macrophage hypothesis of metastasis suggests that metastatic cells arise following fusions of macrophages or bone marrow-derived hematopoietic cells with committed tumor cells. (46)
TNF RECEPTOR SHEDDING
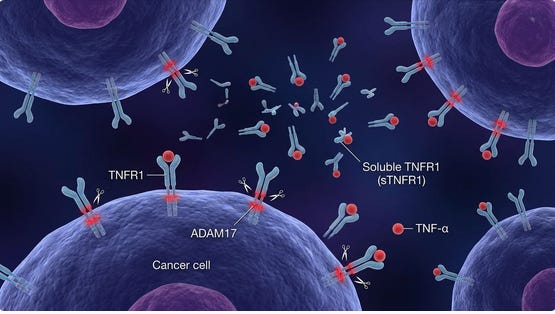
Shedding of TNF receptors (sTNFRs) is a key way tumors and the host regulate TNF signaling, with important implications for aggressive cancers. TNF receptors (primarily TNFR1 and TNFR2) can be proteolytically cleaved from the cell surface by metalloproteases such as ADAM17/TACE, generating soluble TNF receptors (sTNFR1, sTNFR2). This shedding reduces the amount of receptor on the cell surface and releases soluble receptors that can bind circulating TNF, thereby buffering or neutralizing TNF activity and dampening downstream signaling.
Many tumors, including breast cancers, express high levels of ADAM17 and actively shed TNF receptors (sTNFR1/2) and other ligands, reshaping the tumor microenvironment.(61) Soluble TNFRs can sequester TNF in the tumor milieu, limiting TNFR1‑mediated apoptotic or necroptotic killing of tumor cells and blunting the effector function of TNF produced by cytotoxic T cells and NK cells. High expression of TNFR2 on breast cancer cells correlates with larger tumor size, higher stage and grade, and worse overall and disease‑free survival, underscoring the pro‑tumor role of TNFR2‑biased signaling.
The soluble shed receptors act as DECOYS, neutralizing TNF-α before it reaches the tumor. Once these decoys are removed, the body’s own TNF-α is freed to induce tumor lysis and re-activate CD8+ T-cell infiltration. Addressing this via ultrapheresis—physically removing these shed blocking factors—has been shown to be an effective adjunct to restore the body’s innate ability to powerfully induce tumor lysis (see post on potential adjunctive therapies).
I thank Dr W. Campbell Douglass for alerting me to this concept.
Figure 4. Breast cancer cell shedding TNFR1
References
1. de Visser KE, Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41(3):374–403.
2. Mayer S, Milo T, Isaacson A, Halperin C, Miyara S, Stein Y, et al. The tumor microenvironment shows a hierarchy of cell-cell interactions dominated by fibroblasts. Nat Commun. 2023;14(1):5810.
3. Anderson NM, Simon MC. Tumor Microenvironment. Curr. Biol. 2020;30:R921–5.
4. Wang Q, Shao X, Zhang Y, Zhu M, Wang FXC, Mu J, et al. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023;12:11149 – 65.
5. Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, Ostrand-Rosenberg S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc. Biol. 2014;96(6):1109–18.
6. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMEe: Targeting tumor infiltrating myeloid cells to revert cancer progression. Front. Immunol. 2023;9:1977.
7. Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J Med. 2005;353(8):793–802.
8. Cole K, Al-Kadhimi Z, Talmadge JE. Role of myeloid-derived suppressor cells in tumor recurrence. Cancer and Metastasis Reviews. 2023.
9. Ma T, Renz BW, Ilmer M, Koch D, Yang Y, Werner J, et al. Myeloid-Derived Suppressor Cells in Solid Tumors. Cells. 2022;11(2).
10. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–90.
11. Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc. Biol. 2015;98(6):913–22.
12. Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, Chytil A, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70(15):6139–49.
13. Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells. 2020;9(3).
14. Gallego-Ortega D, Ledger A, Roden DL, Law AM, Magenau A, Kikhytyak Z, et al. ELF5 drives lung metastasis in luminal breast cancer through recruitment of Gr1+ CD11b+ myeloid-derived suppressor cells. PLoS Biol. 2015;13:e1002330.
15. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65(8):3044–8.
16. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):1123–31.
17. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70(1):99–108.
18. Sharabi A, tsokos MG, Ding Y, Malek TR, Klatzmann D, Tsokos GC. Regulatory T cells in the treatment of disease. Nature Reviews. 2018;17:823–44.
19. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer. 2020;19(1):116.
20. Raffin C, Vo LT, Bluestone JA. T(reg) cell-based therapies: challenges and perspectives. Nat. Rev Immunol. 2020;20(3):158–72.
21. Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol. Oncol. 2022;15(1):61.
22. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019;110(7):2080–9.
23. Wu T, Wu X, Wang HY, Chen L. Immune contexture defined by single cell technology for prognosis prediction and immunotherapy guidance in cancer. Cancer Commun. (Lond). 2019;39(1):21.
24. Becht E, Giraldo NA, Dieu-Nosjean MC, Sautès-Fridman C, Fridman WH. Cancer immune contexture and immunotherapy. Curr. Opin. Immunol. 2016;39:7–13.
25. Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, et al. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol. Immunol. 2018;15(5):458–69.
26. Giraldo NA, Becht E, Remark R, Damotte D, Sautès-Fridman C, Fridman WH. The immune contexture of primary and metastatic human tumours. Curr. Opin. Immunol. 2014;27:8–15.
27. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev Cancer. 2012;12(4):298–306.
28. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J Cancer. 2013;108(4):914–23.
29. Cassetta L, Pollard JW. Tumor-associated macrophages. Curr. Biol. 2020;30(6):R246–R8.
30. Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. 2020;11:583084.
31. Kumari N, Choi SH. Tumor-associated macrophages in cancer: recent advancements in cancer nanoimmunotherapies. J Exp Clin. Cancer Res. 2022;41(1):68.
32. Heng Y, Zhu X, Lin H, Jingyu M, Ding X, Tao L, et al. CD206(+) tumor-associated macrophages interact with CD4(+) tumor-infiltrating lymphocytes and predict adverse patient outcome in human laryngeal squamous cell carcinoma. J Transl. Med. 2023;21(1):167.
33. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat. Rev Immunol. 2011;11(11):723–37.
34. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016;7:12150.
35. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev Immunol. 2021;21(8):485–98.
36. Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014;105(1):1–8.
37. Liu W, Wang W, Wang X, Xu C, Zhang N, Di W. Cisplatin-stimulated macrophages promote ovarian cancer migration via the CCL20-CCR6 axis. Cancer Lett. 2020;472:59–69.
38. Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer. 2019;18(1):177.
39. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27(4):462–72.
40. Zhao X, Qu J, Sun Y, Wang J, Liu x, Wang F, et al. Prognostic significance of tumor-associated macrophages in breast cancer: a meta-analysis of the literature. Oncotarget. 2017;8(18):30576–86.
41. Yuan X, Zhang J, Li D, Mao Y, Mo F, Du W, et al. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2017;147(1):181–7.
42. Komohara Y, Niino D, Ohnishi K, Ohshima K, Takeya M. Role of tumor-associated macrophages in hematological malignancies. Pathol. Int. 2015;65(4):170–6.
43. Kitano Y, Okabe H, Yamashita YI, Nakagawa S, Saito Y, Umezaki N, et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br. J Cancer. 2018;118(2):171–80.
44. D’Errico G, Alonso-Nocelo M, Vallespinos M, Hermann PC, Alcalá S, GarcÃa CP, et al. Tumor-associated macrophage-secreted 14-3-3 signals via AXL to promote pancreatic cancer chemoresistance. Oncogene. 2019;38(27):5469–85.
45. Gyori D, Lim EL, Grant FM, Spensberger D, Roychoudhuri R, Shuttleworth SJ, et al. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight. 2018;3(11).
46. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013;18:43–73.
47. Fan CS, Chen LL, Hsu TA, Chen CC, Chua KV, Li CP, et al. Endothelial-mesenchymal transition harnesses HSP90α-secreting M2-macrophages to exacerbate pancreatic ductal adenocarcinoma. J Hematol. Oncol. 2019;12(1):138.
48. Wang W, Liu Y, Guo J, He H, Mi X, Chen C, et al. miR-100 maintains phenotype of tumor-associated macrophages by targeting mTOR to promote tumor metastasis via Stat5a/IL-1ra pathway in mouse breast cancer. Oncogenesis. 2018;7(12):97.
49. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell. 2019;35(4):588–602.
50. Debebe A, Medina V, Chen CY, Mahajan IM, Jia C, Fu D, et al. Wnt/b-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene. 2017;36(43):6020–9.
51. Chen Q, Zhang XH, Massagu J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011;20(4):538–49.
52. Yin Z, Ma T, Huang B, Lin L, Zhou Y, Yan J, et al. Macrophage-derived exosomal microRNA-501-3p promotes progression of pancreatic ductal adenocarcinoma through the TGFBR3-mediated TGF-b signaling pathway. J Exp Clin. Cancer Res. 2019;38(1):310.
53. Klimp AH, Hollema H, Kempinga C, van der Zee AG, de Vries EG, Daemen T. Expression of cyclooxygenase-2 and inducible nitric oxide synthase in human ovarian tumors and tumor-associated macrophages. Cancer Res. 2001;61(19):7305–9.
54. Pan B, Ge L, Xun YQ, Chen YJ, Gao CY, Han X, et al. Exercise training modalities in patients with type 2 diabetes mellitus: a systematic review and network meta-analysis. Int. J Behav. Nutr. Phys. Act. 2018;15(1):72.
55. Majety M, Runza V, Lehmann C, Hoves S, Ries CH. A drug development perspective on targeting tumor-associated myeloid cells. FEBS J. 2018;285(4):763–76.
56. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–90.
57. Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci U. S. A. 2014;111(30):E3053–E61.
58. McCarty OJ, Mousa SA, Bray PF, Konstantopoulos K. Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood. 2000;96(5):1789–97.
59. Heinmoller E, Weinel RJ, Heidtmann HH, Salge U, Seitz R, Schmitz I, et al. Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J Cancer Res Clin. Oncol. 1996;122(12):735–44.
60. Grignani G, Pacchiarini L, Ricetti MM, Dionigi P, Jemos V, Zucchella M, et al. Mechanisms of platelet activation by cultured human cancer cells and cells freshly isolated from tumor tissues. Invasion Metastasis. 1989;9(5):298–309.
Nice breakdown. One critical mechanism of immune evasion that deserves a spot in this playbook is the shedding of TNF RECEPTORS (sTNFRs). Many aggressive tumors utilize enzymes to 'shed' their surface receptors into the serum. This creates a systemic 'sink' that prevents the immune system from inducing apoptosis. The soluble, shedded receptors act as DECOYS, neutralizing TNF-α before it reaches the tumor. Once these decoys are removed, the body's own TNF-α is freed to induce RAPID tumor lysis and re-activate CD8+ T-cell infiltration. It’s a powerful way to turn a tumor’s own defense mechanism against itself. Addressing this via ultrapheresis—physically removing these shed blocking factors—has been shown by Letz and others to be an effective adjunct to restore the body's innate ability to powerfully induce tumor lysis. It’s a mechanical solution to a biochemical shield and fits the model of pregnancy in most animals (and all mammals). Founational citation on “subtractive immunotherapeutic apheresis: https://immunicom.com/news/aacr-annual-meeting-2023/
Diagnosed SLE here. Currently going through ovarian cancer treatment. Would it be fair to say that there should be more collaboration between rheumatologists and oncologists at the start of Lupus diagnoses to determine if cancer is involved should be more common practice? None of my rheumatologists looked at cancer as a factor, although I had markers in my blood that indicated there could be an issue. My cancer wasn’t discovered until my obgyn paid attention to those numbers and other symptoms I reported.